

Abstract
Murine models of Acute Lymphoblastic Leukaemia (ALL) suggest relapse arises not from intrinsic chemoresistance by genetically distinct cells, but from a subset of cells protected within a specific niche.
To confirm the existence of such a niche in patients with ALL and elucidate the mechanism by which stromal cells (MSC) protect ALL cells we isolated MSC from bone marrow of 70 B-ALL patients enrolled on the UKALL14 trial. Immunostaining for f-actin, gene expression profiling and cytokine/chemokine quantification showed that a significant proportion of bone marrow specimens, especially after treatment with a cytarabine (AraC)-containing block, contained MSC with an activated phenotype, analagous to cancer associated fibroblasts (ALL-CAF).
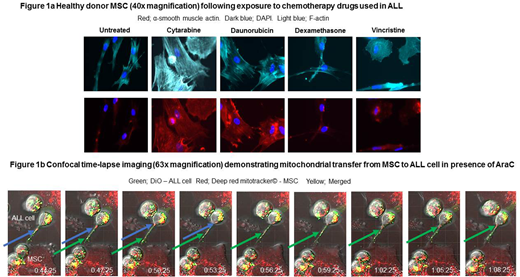
We demonstrated that primary ALL cells, ALL cell lines, AraC and daunorubicin (but not vincristine (VCR) or dexamethasone (dex)) generated ALL-CAF de novo from both healthy donor MSC and the MSC cell line HS27a. Notably, the chemotherapy drugs induced distinct morphological changes and differential alpha-smooth muscle expression (figure 1a).
Control of oxidative stress via modification of reactive oxygen species (ROS) presented a potential unifying explanation for ALL-CAF generation by both ALL cells and chemotherapy. Using flow cytometry we demonstrated that AraC significantly increased ROS in the B-ALL cell line, SEM, in monoculture but in co-culture with HS27a, ROS was significantly lowered and was not impacted by AraC. The MSC co-culture-mediated reduction in ROS in co-culture corresponded to a significant reduction in cell death (10.5% vs 36%, p = 0.0001). By contrast, VCR did not impact ROS significantly and Dex reduced it. Both were significantly more effective than AraC at inducing SEM cell death in co-culture (VCR 20.2% vs AraC 10.5%, p = 0.0003; Dex 39.1% vs AraC 10.5%, p = 0.0007), despite inducing the same degree of cell death in monoculture.
We hypothesised that mitochondrial transfer between ALL-CAF and B-ALL cells could provide a generalised mechanism to overcome the deleterious impact of cell-intrinsic and chemotherapy-driven ROS in B-ALL cells. A 'mitotracker' flow cytometry assay showed differential mitochondrial transfer from HS27a to B-ALL cells, in proportion to the baseline ROS levels. We confirmed that mitochondria could also be transferred from healthy donor MSC co-cultured with primary patient ALL cells. Furthermore, AraC, but not VCR or Dex, significantly enhanced mitochondrial transfer, and did so in a dose-dependent manner. To rule out passive transfer of dye, we used the murine stromal line MS5 as an alternative mitochondria donor to SEM cells. Murine mitochondrial, but not nuclear, DNA was clearly seen in flow-sorted SEM cells after co-culture with MS5, at baseline and at higher levels after AraC therapy. We also directly visualised the transfer of mitochondria along tunnelling nanotubes (TNT) by time-lapse confocal imaging (figure 1b). To confirm that mitochondrial transfer was essential in MSC 'rescue' of ALL cells, we generated HS27a cells deficient in mitochondria following prolonged culture with low dose ethidium bromide. The mitochondrially-deficient cells retained viability as well as the ability to become ALL-CAF but were clearly defective in their ability to rescue SEM ALL cells from AraC induced cell death.
To confirm the functional impact of mitochondrial transfer via TNT, we used actin inhibitor latrunculin B (LatB) and the microtubule damaging agent nocodazole which both significantly blocked the phenomenon. Both LatB and nocodazole significantly restored AraC-related cytotoxicity. Colchicine, another microtubule damaging agent had a similar impact to nocodazole. VCR completely overcame the protective impact of HS27a on AraC cytotoxicity and was additive with AraC in the co-culture system.
We have shown that CAF-like MSC provide support to ALL cells under oxidative stress by mitochondrial transfer via TNT. This is disrupted by microtubule damaging agents and conditions provoking their formation are mitigated by Dex, both mainstays of ALL therapy. Our data may explain the ineffectiveness of ROS-inducing chemotherapy at eradicating disease at the niche and provides an explanation of why low dose, microtubule damaging agents such as VCR used in maintenance therapy are effective in ALL. Our findings have immediate implications for the design and scheduling of current combination chemotherapies for ALL.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal